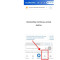
Patofisiologi Adenoma Pituitari
Patofisiologi adenoma pituitari atau adenoma hipofisis belum sepenuhnya dipahami, tetapi sebagian besar bersifat sporadis. Adenoma pituitari merupakan neoplasma epitel jinak yang terdiri dari sel adenohipofisis. Tumor pituitari ganas primer sangat jarang dijumpai.[1,5]
Sekilas Anatomi Kelenjar Pituitari
Kelenjar pituitari terdiri dari dua lobus, yaitu lobus anterior (adenohipofisis) dan lobus posterior (neurohipofisis). Adenohipofisis terdiri dari tiga bagian yaitu pars distalis (lobus anterior atau pars glandularis), pars intermedia (lobus intermedia), dan pars tuberalis (pars infundibularis).[1-4]
Adenohipofisis ini meregulasi beberapa proses fisiologi termasuk stres, pertumbuhan, dan reproduksi. Adenohipofisis menghasilkan enam jenis hormone, yaitu thyroid stimulating hormone (TSH), follicle stimulating hormone (FSH), growth hormone (GH), luteinizing hormone (LH), corticotropin, dan prolaktin.[1-4]
Neurohipofisis berasal dari perkembangan otak dan juga terdiri dari tiga bagian yaitu infundibulum, proyeksi ke bawah ke arah tuber cinereum berbentuk terowongan; tangkai hipofisis; lobus posterior atau pars nervosa. Neurohipofisis mensekresikan vasopresin dan oksitosin. Vasopresin juga dikenal sebagai anti diuretik hormon (ADH) karena fungsinya yang meretensi cairan di ginjal.[1-4]
Mutasi Genetik
Perkembangan tumor pituitari umumnya merupakan suatu proses monoklonal dengan beberapa faktor penyebab. Sifat monoklonal sebagian besar tumor pituitari menunjukkan bahwa tumor tersebut muncul dari sel pituitari yang bermutasi. Namun, mekanisme patofisiologis maupun molekuler yang menyebabkan terbentuknya adenoma pituitari masih belum diketahui.[1,5]
Mutasi genetik jarang dijumpai pada adenoma pituitari. Peran mutasi genetik dilaporkan dalam satu laporan kasus yang menunjukkan bahwa pasien dengan tumor pituitari dari 4 keluarga Irlandia memiliki mutasi gen yang sama dengan pasien dari abad ke-18 yang mengalami gigantisme yang dimediasi tumor pituitari.[1,5]
Mutasi pada Gen Multiple Endocrine Neoplasia Type 1 (MEN1)
Gen MEN1 adalah gen penekan tumor, sehingga mutasi gen ini menyebabkan pembentukan tumor di kelenjar paratiroid, pankreas, dan pituitari. Pada pasien yang tidak terdeteksi mutasi pada gen MEN1, mungkin memiliki mutasi di area regulasi, tidak diterjemahkan, atau intron yang masih terus diteliti. Delesi atau rearrangement bruto lainnya mewakili 1‒3% dari mutasi MEN1, dan dapat dideteksi dengan analisis Southern blot atau multiplex ligation-dependent probe amplification (MLPA).[1,6]
Mutasi pada Gen Multiple Endocrine Neoplasia Type 4 (MEN4)
Mutasi pada gen MEN4 disebabkan oleh mutasi pada gen cyclin-dependent kinase inhibitor 1B (CDKN1B/12p13.1-p12) encoding p27, yang muncul dengan tumor pituitari, hiperparatiroidisme, tumor testis, dan tumor neuroendokrin serviks. Gen CDKN1B merupakan cyclin-dependent kinase inhibitor yang bertindak sebagai regulator negatif dari perkembangan siklus sel.[1,7]
Mutasi pada Gen Carney Complex (CNC)
Pada gen CNC, terdapat mutasi germline dari gen penekan tumor PRKAR1A yang mengarah ke penyakit adrenokortikal nodular berpigmen primer (PPNAD), tumor testis, nodul tiroid, hiperpigmentasi pada kulit, akromegali, Schwannoma melanositik, dan miksoma jantung. Sampai sekarang, dua lokus telah terlibat secara pasti dalam genetika CNC, yaitu gen CNC 1 dan CNC 2.[1,8]
Gen CNC 1 terletak di 17q22-24, yang mengkode subunit regulator (R1a) dari protein kinase A (PRKAR1A), dan bertanggung jawab atas 2/3 kasus adenoma pituitari. Sedangkan gen CNC 2 pada lokus 2p16 belum teridentifikasi.[1,8]
Mutasi pada Gen Familial Isolated Pituitary Adenomas (FIPA)
Mutasi Aryl Hydrocarbon Receptor-Interacting Protein (AIP) dilaporkan pada masa remaja atau dewasa awal pada sekitar 15% dari semua FIPA. Tumor ini biasanya bersifat agresif dan merupakan jenis adenoma yang mensekresi growth hormone.[1,11]
Jenis FIPA kedua yang dikarakterisasi secara genetik adalah X-linked acrogigantism (XLAG) yang disebabkan oleh duplikasi GPR101. XLAG merupakan gangguan yang sangat penetran, dikaitkan dengan hiperplasia pituitari atau adenoma yang mengakibatkan kelebihan hormon pertumbuhan dengan onset pada masa bayi, biasanya terkait dengan hiperprolaktinemia. Kebanyakan individu dengan XLAG memiliki perubahan genetik mosaik somatik de novo yang tidak diwariskan dari orang tua.[1,11]
Mutasi Pada Gen Guanine Nucleotide Binding Alpha Stimulating (GNAS1)
Mutasi pada gen GNAS1 saat masa embrionik dapat memicu sindrom McCune-Albright. Pada sindrom McCune-Albright, lesi kulit dan displasia fibrosa poliostotik terjadi dengan endokrinopati yang hiperfungsi. Sindrom ini terjadi akibat pengaktifan mutasi (mutasi somatik) subunit alfa protein Gs dan melibatkan jaringan yang responnya terhadap sinyal hormonal dimediasi oleh adenylate cyclase.[10,11]
Tumor pituitari yang paling umum pada sindrom McCune-Albright adalah somatotropinoma, yang menyebabkan akromegali. Sebagian besar somatotropinoma pada kasus akromegali sporadis mengandung mutasi yang sama.[10,11]